
Jantarika Kumar Arora, Louisa K James, Varodom Charoensawan
Abstract
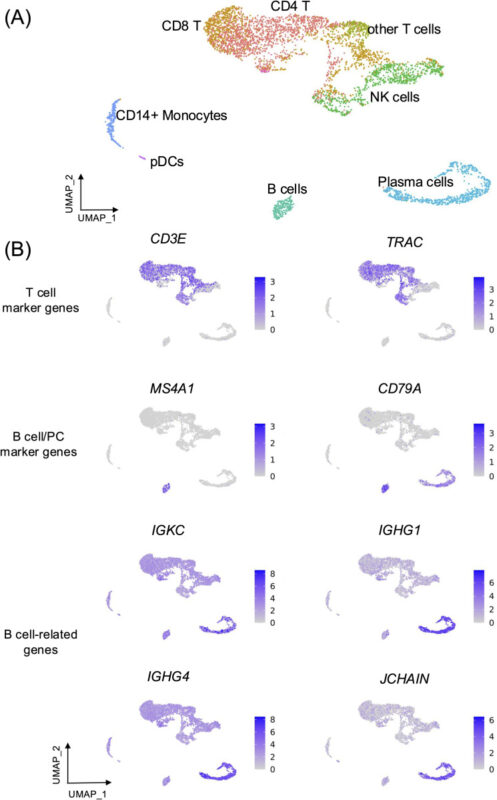
Droplet-based single-cell RNA sequencing (scRNA-seq) frequently encounters significant challenges from contamination of cell-free mRNAs, known as “ambient mRNAs”, which can substantially distort single-cell transcriptome data interpretation to a large extent. In this study, we investigate the impact of ambient mRNA contamination on differential gene expression and biological pathway enrichment analyses, using two independent scRNA-seq datasets: ten peripheral blood mononuclear cells (PBMCs) samples from dengue-infected patients and forty-two scRNA-seq samples of human fetal liver tissues. We apply two independent ambient mRNA correction approaches – CellBender (automate correction) and SoupX (using a predefined set of potential ambient mRNA genes). We demonstrate that ambient mRNA transcripts appear among differentially expressed genes (DEGs), subsequently leading to the identification of significant ambient-related biological pathways in unexpected cell subpopulations before ambient mRNA contamination correction. In contrast, after suitable correction, we observe a reduction in ambient mRNA expression levels, resulting in improved DEG identification and leading to the highlight of biologically relevant pathways specific to cell subpopulations. Our study underscores the importance of understanding and applying appropriate corrections for ambient mRNA contamination to enhance the reliability and accuracy of scRNA-seq data analyses, thereby improving the robustness of data interpretation in droplet-based scRNA-seq datasets.
